Role of NADPH oxidases during myocardial ischemia-reperfusion
Oxidative stress, the presence of reactive oxygen species (ROS) in excess of the antioxidant capacity in the heart induces myocardial damage, accumulation of which leads to ischemic heart disease and heart failure. NADPH oxidase Nox 2 and 4 are the major sources of O2− and H2O2 in the heart and play a crucial role in the regulation of growth and death in cardiomyocytes. Both Nox2 and Nox4 are upregulated in response to ischemia-reperfusion (I/R), thereby contributing to ROS production and consequent myocardial injury. Suppression of either one of them can reduce ROS and I/R injury in the heart. However, a minimum level of ROS production by either Nox2 or Nox4 is essential for the activation of HIF-1α and inhibition of PPARα during I/R, such that the combined suppression of both Nox2 and Nox4 exacerbates myocardial I/R injury. Thus, either excessive activation or suppression of Noxs below physiological levels can induce cardiac injury. Noxs are the only known enzymes whose sole biological function is to purposefully produce O2− or H2O2, and they are major sources of reactive oxygen species (ROS) in cardiovascular cell types. Nox2 and Nox4 are abundantly expressed in cardiomyocytes and play a crucial role in the development of cardiac injury and remodeling. It was shown recently that hypertrophic stimuli induce upregulation of Nox4, thereby leading to increased production of ROS, mitochondrial dysfunction and apoptosis in cardiomyocytes. Cardiac hypertrophy and dysfunction in response to pressure overload was significantly attenuated in cardiac-specific Nox4 knockout (KO) mice, and this was accompanied by the preservation of mitochondrial function. Although Nox2 is involved in cardiac remodeling, including cardiomyocyte hypertrophy, apoptosis and interstitial fibrosis, after permanent coronary ligation, Nox2 is not essential for the development of cardiac hypertrophy after pressure overload. In contrast, Nox1 is expressed mainly in the endothelial cells in the heart and is involved in endotoxin induced cardiomyocyte apoptosis. These findings indicate that Noxs have isoform-specific functions in the heart during cardiac remodeling. Investigating the function of Nox2 and Nox4 in the heart using systemic Nox4 KO (sNox4 KO) and systemic Nox2 KO (sNox2KO) mice, both types of mice exhibited similar levels of reduction in ROS production and attenuation of the infarct size after I/R, indicating that the ROS produced by Nox2 and Nox4 at distinct subcellular localizations contribute equally to the overall increase in ROS and myocardial injury in response to I/R. I/R injury was also reduced in cardiac-specific Nox4 KO mice to a similar extent as in sNox4 KO mice, suggesting that the Nox4 in cardiomyocytes, rather than in non-myocytes, plays a dominant role in mediating I/R injury. A recent report suggests that abolishing Nox activity through overexpression of DN-Nox, which broadly suppresses Nox isoforms, leads to a ‘markedly reduced state’ in the heart, which is characterized by low NAD(P)+/NAD(P)H and high GSH/GSSG ratios. This most likely occurs through decreased consumption of NAD(P)H due to the suppression of Noxs. Since Nox4 is localized on mitochondrial membranes and consumes NADH, suppression of Nox4 could induce accumulation of NADH in mitochondria. Interestingly, when NADH accumulates in the presence of slow flow in the mitochondrial electron transport chain (ETC) during ischemia, direct transfer of electrons to oxygen takes place, resulting in excessive production of superoxide and severe irreversible damage of the heart. Thus, Noxs regulate the redox state of cardiomyocytes not only through direct production of ROS but also through consumption of NAD(P)+/NAD(P)H. Mitochondrial ROS derived from Nox4 oxidize the cysteines of aconitase-2 and citrate synthase, leading to mitochondrial dysfunction and apoptosis in cardiomyocytes during aging and heart failure. In the ER, Nox4 mediates autophagy during glucose deprivation in cardiomyocytes.
I/R-induced upregulation of Nox2 and Nox4 induces O2− production in both plasma and intracellular membranes. O2− is rapidly dismutated to H2O2, which is diffusible and membrane permeable. Since PHD2, one of the critical targets of ROS mediating metabolic adaptation during I/R, is localized primarily in the cytosol, both Nox2 and Nox4 can contribute equally to the inactivation of PHD2 and stabilization of HIF-1α (Fig. 9).
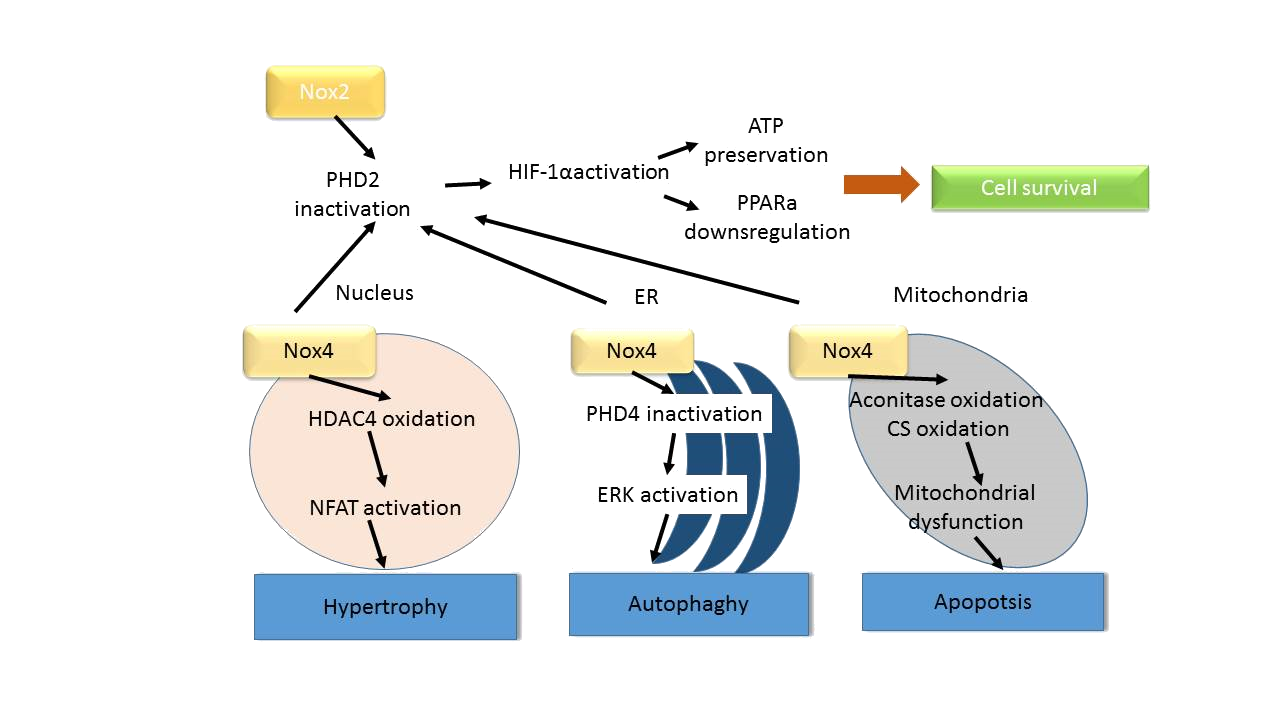
Figure 9: Schematic representation of the signaling pathways controlling growth and death of cardiomyocytes that are regulated by Nox isoforms. Nox2 and Nox4 are localized in different subcellular compartments and regulate signaling mechanisms locally or globally to affect metabolism, hypertrophy, autophagy and apoptosis
Another important issue to consider when discussing the function of Noxs in the heart is that the function of Nox appears to be cell type-dependent. For example, cardiac-specific deletion of Nox4 inhibits pressure overload-induced cardiac hypertrophy and left ventricular dysfunction. In contrast, systemic deletion of Nox4 exacerbates cardiac remodeling after pressure overload. A side-by-side comparison, using mice derived from the same flox mice to generate cardiac and systemic KO mice, showed that cardiac and systemic Nox4 KO mice exhibited distinct cardiac phenotypes. Cardiac hypertrophy in response to pressure overload was significantly attenuated in cardiac-specific Nox4 KO mice but not in systemic Nox4 KO mice. Furthermore, interstitial fibrosis and diastolic dysfunction were exacerbated in systemic Nox4 KO mice. It was speculated that Nox4 in cardiomyocytes promotes cardiac dysfunction and consequent hypertrophy, whereas Nox4 in non-myocytes protects the heart from fibrosis and diastolic dysfunction. Interestingly, Nox4 exhibits salutary effects against pathologically relevant stresses in some cell types. For example, Nox4 in endothelial cells is protective against angiotensin II-induced endothelial dysfunction, in contrast to Nox1 and Nox2. Although Nox may be an ideal target for clinical therapy to prevent oxidative stress, it must be taken into consideration that Noxs are also involved in physiological and adaptive cellular functions. Unconditional administration of antioxidants such as Vitamin E as a treatment for cardiovascular disease has not been successful, possibly due to the broad suppression of ROS.
As a conclusion, Nox isoforms play an important role in mediating oxidative stress and myocardial injury after pressure overload and I/R. In addition suppressing Nox isoforms can effectively prevent excessive activation of ROS in a stimulus-specific manner. In the context of I/R, however, a certain amount of ROS derived from either Nox2 or Nox4 is required to protect the heart against I/R via powerful metabolic adaptations triggered by HIF-1α-PPARαdependent mechanisms. Thus, although either Nox2 or Nox4 can be targeted for the treatment of I/R, maintenance of a minimum level of Nox activity is needed to preserve the physiological function of Nox.